In the previous post, we mentioned how deep-learning and generative approaches are revolutionizing genomics and single-cell studies.
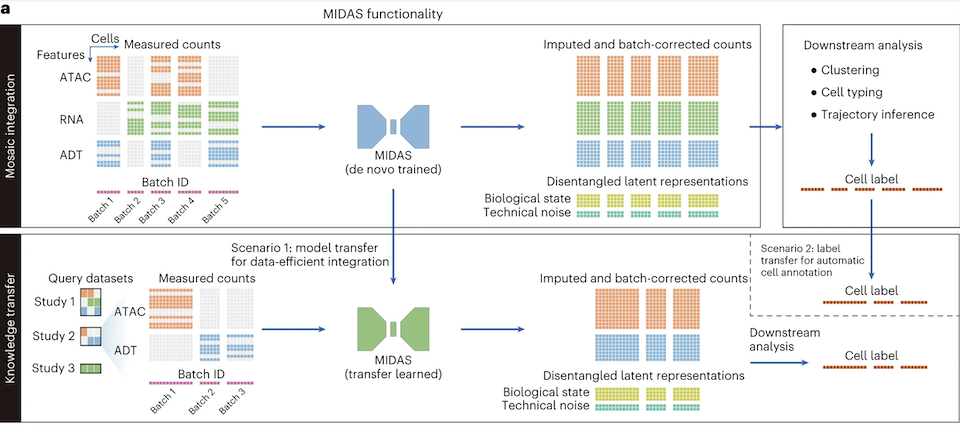
Today, we talk about another model that has a distinct purpose called MIDAS (mosaic integration and knowledge transfer). MIDAS is designed to handle and improve data integration across different single-cell modalities, e.g., RNA-Seq, ATAC-Seq, and CITE-seq or ADT (antibody-derived tags).
That sounds fancy. But what is the purpose of this approach?
Well, single-cell studies are often limited by data sparsity, i.e., zeros in the count matrices. These so-called “gaps” in the data can dramatically reduce the biological insights related to a specific disease or condition. In this context, MIDAS shines as a way to effectively integrate multimodal data or even generate data modality layers for improving downstream analysis. Yes, I said generate.
In this study, the authors demonstrate how the method performs to fulfill this data mosaic. Not surprisingly, the generated layers seem to improve cell clustering and highlight cellular heterogeneity at a fine-grained level in a PBMC dataset. Further, the authors pushed the boundaries and mentioned that MIDAS can even help detect rare proteins in B cells (e.g., CD20). Other cell markers were also brought to attention such as CD3 and CD4 (T-cell markers) observed in two B subclusters. Intriguing.
Anyhow, if you are a researcher and you facing issues analyzing your single-cell project - cough, cough low cell counts - I would suggest adding MIDAS-like methods to your workflow.
Yet, remember generative models are still biased and sometimes can lead to misinterpretations. Maybe a before-and-after imputation approach could be a nice way to go… I leave it to you, dear bioinformatician.